

视神经脊髓炎
发布时间:2021-10-12 点击量:1660
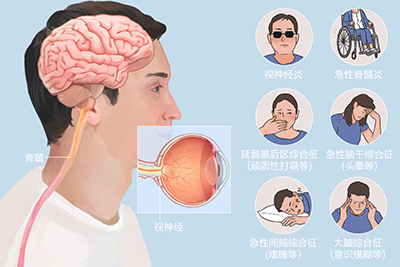
视神经脊髓炎(neuromyelitis optica,NMO)是视神经与脊髓同时或相继受累的急性或亚急性脱髓鞘病变。该病由Devic(1894)首次描述,其临床特征为急性或亚急性起病的单眼或双眼失明,在其前或其后数日或数周伴发横贯性或上升性脊髓炎,后来本病被称为Devic病或Devic综合征。资料显示NMO占所有脱髓鞘病的1%-22%,在西方国家比例偏低,在非高加索人比例偏高。
2018年5月11日,国家卫生健康委员会等5部门联合制定了《第一批罕见病目录》,视神经脊髓炎被收录其中。
视神经脊髓炎分类
(1)单时相病程NMO:占10-20% [2] ,欧洲相对多见,病变仅限于视神经和脊髓,视神经炎多为双侧同时受累与脊髓炎同时或相近(1 月内)发生,神经功能障碍常较复发型 NMO 重。但是生存期较长。 [3]
(2)复发型 NMO:占80%-90% [3] ,亚洲相对多见,初期多表现为单纯的孤立视神经炎或孤立脊髓炎,仅约10%的患者首次发病视神经脊髓同时受累 [2] 。
(3)进展型NMO :少见。
视神经脊髓炎谱系疾病
有一些发病机制与NMO类似的非特异性炎性脱髓鞘病,其NMO-IgG阳性率亦较高。 Wingerchuk 将其归纳并提出了视神经脊髓炎谱系疾病(neuromyelitis optica Spectrum disorders,NMOSDs)概念 [4] 。 2010年欧洲神经病学联盟(EFNS)将 NMOSDs明确定义,特指一组潜在发病机制与NMO 相近,但临床受累局限,不完全符合NMO诊断的相关疾病 [5] 。
(1)2010 EFNS NMOSDs [5]
①受累部位局限的类型,如长节段横断性脊髓炎(1ongitudinally extensive transverse myelitis,LETM)、复发性孤立性视神经炎(recurrent isolated optic neuritis,RION)和双侧视神经炎(bilateraloptic neuritis,B0N)。
②在器官特异性或非器官特异性自身免疫疾背景下发生的 NMO。
③伴有症状性或无症状性脑内病灶的不典型病例。
④亚洲国家的视神经髓型 MS(optic-spinal MS,OSMS)。
(2)2007 Wingerchuk NMOSDs
① NMO。
② 病变限定于视神经和脊髓。
a) 特发性单时相或复发性长节段横惯性脊髓炎(MRI病灶≥ 3个椎体节段)。
b)视神经炎:复发性视神经炎或同时发生的双侧视神经炎。
③亚洲类型的视神经脊髓型多发性硬化(OSMS)。
④视神经炎或长节段横惯性脊髓炎合并自身免疫性疾病。
⑤ 视神经炎或脊髓炎合并 NMO 特征的颅内病灶(下丘脑、胼胝体、脑室周边及脑干)。
一项研究表明,横贯性脊髓炎NMO-IgG阳性患者有55%在1年内脊髓炎复发或发展为NMO。
病因及发病机制
语音
病因尚不清楚。
AQP4是中枢神经系统主要的水通道蛋白,位于星形胶质细胞的足突上,AQP4是NMO-IgG的主要目标,这解释了NMO的病灶主要位于视神经及脊髓。AQP4抗体通过血脑屏障中可通过的部分进入中枢神经系统,立即遇到星形胶质细胞并导致细胞依赖的细胞毒性反应,星形胶质细胞足突被NMO-IgG和补体降解,继而活化的巨噬细胞、嗜酸性粒细胞及中性粒细胞一起产生细胞因子、氧自由基等造成血管和实质损伤,最终导致包括轴索和少突胶质细胞在内的白质和灰质的损伤。
家族性NMO病例少见,在所有确诊NMO中少于3%。人白细胞抗原DPB1*0501(亚洲人群)及 DRB1*0301(高加索人群)与NMO易感性相关。说明遗产因素在NMO发病中有一定作用。
病理生理
语音
NMO病变主要累及视神经、视交叉和脊髓(胸段与颈段)。其病理改变与NMO患者的生存期有关。
脊髓病理
(1)大体病理:一般多个脊髓节段受累,通常可从胸髓波及至颈髓或腰髓。早期致死性病例可见脊髓发生肿胀和软化;生存期较长的患者,脊髓可发生皱缩。
(2)显微镜下病理:脊髓肿胀软化部位镜下可见病变累及脊髓灰质和白质,坏死组织呈灶状或融合成片状,可见小的囊腔形成,轴索和神经细胞丢失,中性粒细胞浸润,毛细血管增生,可见血管周围淋巴细胞袖套样浸润;其他部位的脊髓可见散在或融合成片的脱髓鞘改变。脊髓皱缩部位可见空腔形成,间质明显增生,上行及下行神经纤维束Wallerian变性。
视神经病理
视神经炎症包括淋巴细胞、巨噬细胞、单核细胞浸润,及血管炎症。长时间后可见坏死及空洞形成,血管内皮细胞增殖,神经胶质细胞增生或损失,视神经及视交叉脱髓鞘。逆行性轴索损伤导致视网膜神经纤维层丢失。
其他
部分NMO患者中枢神经系统的其他部位,如脑干、脑室周围、半卵圆中心白质等可出现类似经典MS样的脱髓鞘病灶。
临床表现
语音
有限流行病学资料显示,NMO的患病率是0.3-4.4/100,000,年发病率是0.05–0.4/100,000。男女均可发病,单时相 NMO 男女患病比率相等,复发型 NMO 女性发病显著高于男性 [2] ,女性/男性患病比率约为 9-12:1。平均发病年龄 30-40岁,约 10%的 NMO 患者发病年龄小于 18岁。
NMO 主要有视神经和脊髓两大组症候,部分患者合并有脑干损害症状。大约一半的病人以孤立视神经炎起病,其中20%的病人双侧视神经炎;一半的病人以孤立的脊髓炎起病;10%的病人视神经及脊髓同时受累。
视神经症候
眼痛、视力下降或失明、视野缺损。可单眼、双眼间隔或同时发病。
脊髓症候
以横惯性脊髓损害较为多见,包括有脊髓相应病变平面以下传导束型深浅感觉、运动障碍及膀胱直肠功能障碍,神经根性疼痛、痛性痉挛,Lhermitte征,高颈段受累者可出现呼吸肌麻痹症候。
脑干症候包括
①顽固性呃逆、恶心、呕吐等延髓颈髓交界区受累症状,此表现在NMO 中相对特异,有些病例为唯一首发表现。②间脑病变可出现嗜睡、困倦、低钠血症等。
辅助检查
语音磁共振成像(MRI)
1.头颅 MRI:许多NMO患者有脑部病灶,大10%约的NMO患者脑部病灶与MS一致。其分布多与AQP4高表达区域相一致,而不符合MS的影像诊断标准。特征性病灶位于下丘脑、丘脑、三脑室、导水管、桥脑被盖及四脑室周围。延髓病变,常与颈髓病灶相延续。病变往往不强化。
此外假瘤样脱髓鞘和可逆性后部白质脑病亦可见于患者。
2.眼部 MRI:急性期可见视神经增粗、肿胀,呈长T1、长T2信号,可见“轨道样”强化。通常双侧视神经均有异常,视交叉及视觉传导通路上可见异常。
3.脊髓 MRI:病变常累及 3 个或 3 个以上椎体节段,为NMO 最具有特异性的影像表现。NMO 以颈段或颈胸段同时受累最为多见,病变可向上延伸至延髓下部。病变多位于脊髓中部,累及大部分灰质和部分白质。急性期多伴有脊髓肿胀并可见强化。疾病后期部分病例脊髓变细、萎缩、中心空洞形成。
脑脊液检查
急性期脑脊液中性粒细胞和嗜酸性粒细胞增多较常见,约13-35%患者细胞数大于 50/mm3。46-75%患者脑脊液蛋白升高。小于30%的NMO患者脑脊液寡克隆区带可阳性。
血清 NMO-IgG
NMO-IgG是NMO的免疫标志物,是鉴别NMO与MS的重要参考依据之一,需反复检测。此外,NMO 患者 NMO-IgG 强阳性其复发可能性较大,其滴定度有可能作为复发与治疗疗效的评价指标。实验方法不同阳性率不同,NMO患者血清 NMO-IgG阳性率大约50-75%。最敏感的方法是细胞转染免疫荧光法。
血清自身抗体
约 40-60%的 NMO 患者可伴有其他自身免疫疾病抗体阳性。如抗核抗体、抗 SSA/SSB抗体、抗心磷脂抗体,甲状腺相关抗体,乙酰胆碱受体抗体等。
神经眼科检查
1.视敏度:80%以上NMO患者仅为 20/200 或更差,超过30%的患者无光感;第一次发病后30%患者的视力低于20/200;病程 5 年以上的 NMO 患者,有一半患者单眼视敏度低于 20/200,其中 20%的患者为双眼视敏度降低。
2.视野检查: NMO患者可有中心及外周视野缺损。
3.视网膜厚度(OCT):NMO 患者视网膜神经纤维层(RNFL)明显缺失,平均减少厚度约为30-40UM,而MS平均减少厚度为20-30 UM。Ratchford等人发现NMO相关神经炎首次发作时RNFL减少31 UM,以后每次发作减少10 UM。RNFL与视力、视野、功能缺损、疾病进程相关。 平均RNFL低于70 UM时将会发生失明。
4.视觉诱发电位(VEP):多数患者有 VEP 异常,主要表现为 P100 潜时延长、波幅降低或P100引不出。部分患者可发现亚临床病灶。
诊断及鉴别诊断
语音诊断
我国专家推荐使用 2006年Wingerchuk 修订的 NMO 诊断标准 [6] ,其敏感性和特异性分别为 87.5%和 83.3%。如下:
(1)必备条件(下列每项至少有 1次发作)①视神经炎 ②横贯性脊髓炎。
(2)支持条件(至少两项)①MRI:正常或病变不符合多发性硬化影像学诊断标准。②脊髓 MRI:病灶超过 3个脊椎节段。③血清 NMO-IgG阳性。
具备必要全部条件和支持条件中的2条,即可诊断NMO。
鉴别诊断
1.多发性硬化 鉴别要点如下
|
鉴别特点
|
NMO
|
MS
|
|
流行病学
|
||
|
男:女比例
|
1:(5-10)
|
1:(2-3)
|
|
发病年龄
|
30-40岁
|
20-40岁
|
|
病程
|
||
|
类型
|
复发型,早期复发率高
|
复发-缓解型
|
|
病情
|
严重,不完全恢复
|
较轻,恢复较好
|
|
永久性残疾
|
和复发相关
|
通常在进展期
|
|
临床表现
|
||
|
受累部位
|
视神经和脊髓
|
视神经、脊髓、小脑、脑干、大脑半球
|
|
脊髓炎
|
ACTM,累及延髓导致顽固性恶心、呃逆或呼吸衰竭
|
APTM
|
|
视神经炎
|
严重,双侧同时或相继快速发生
|
轻或中度
|
|
脑部症状
|
可以(脑病,下丘脑功能障碍)
|
常有(复视、核间性眼肌麻痹、偏身感觉障碍或无力)
|
|
辅助检查
|
||
|
脊髓MRI
|
超过3个或更多脊髓阶段、灰质中央或整个脊髓横断面、伴肿胀和扎增强
|
少于2个脊髓阶段、非对称性偏心分布,累及脊髓后部、没有或很少肿胀
|
|
脑部MRI
|
MRI正常或不符合MS特征(环绕脑室管膜周围区域)融合、线样病灶
|
脑室旁、近皮层、幕下、长轴垂直于脑室壁的圆形结构
|
|
脑脊液
|
细胞增多,大于50X10∧6/L、中性粒细胞为主、蛋白升高常见、OCB阴性,IgG指数升高少见
|
细胞增多不常见、单核细胞为主、蛋白升高不常见、OCB阴性,IgG指数升高
|
|
血清NM0-IgG
|
常见
|
罕见
|
|
合并自身抗体或
|
常见
|
罕见
|
(2)视神经炎:多损害单眼,而NMO常两眼先后受累,并有脊髓病损或明显缓解-复发。
(3)急性脊髓炎:起病急,瘫痪呈横贯性脊髓损害表现,病程中无缓解复发,也无视神经损害表现。
(4)Leber 视神经病、亚急性坏死性脊髓病、亚急性联合变性、脊髓硬脊膜动静脉瘘、梅毒性视神经脊髓病、脊髓小脑性共济失调、遗传性痉挛性截瘫、脊髓肿瘤、脊髓血管病、热带痉挛性瘫痪、肝性脊髓病,某些结缔组织病,如系统性红斑狼疮、白塞氏病、干燥综合症、系统性血管炎等伴发的脊髓损伤,也应注意与 NMO 相鉴别。
治疗
语音急性发作/复发期治疗
1.糖皮质激素
最常用一线治疗方法,抑制炎症反应,促进白细胞凋亡及抑制多形核白细胞迁移,可减轻疾病的炎性活动及进展,保护神经功能。应用原则是:大剂量,短疗程,减药为先快后慢,后期减至小剂量长时间维持。
具体方法:甲泼尼龙1g,静滴1/日×3-5天, 500mg静滴1/日×3天, 240mg静滴 1/日×3天,120mg 静滴 1/日×3 天,60mg 口服后缓慢阶梯减量至小剂量长时间维持。对激素依赖性患者,激素减量过程要慢,可每周减5mg,至维持量(每日 2-4片),小剂量激素须长时间维持。
激素有一定副作用,电解质的紊乱,血糖、血压、血脂异常,上消化道出血,骨质疏松,股骨头坏死,脂肪重新分布等。激素治疗中应注意补钾、补钙,应用抑酸药。
2.血浆置换(plasma exchange,PE)
与血浆中的自身抗体、补体及细胞因子等被清除有关。对于症状较重及糖皮质激素治疗无效的患者有一定效果。用激素冲击治疗无效的 NMO 患者,用血浆置换治疗约 50%仍有效。经典治疗方案通常为在 5-14 天内接受 4-7 次置换,每次置换约 1-1.5 倍血浆容量。一般建议置换 3-5 次,每次血浆交换量在 2-3L,多数置换 1-2 次后见效。
3. 静脉注射大剂量免疫球蛋白 [7] (Intravenous immunoglobulin, IVIg)
可用于急性发作,对激素反应差的患者。用量为0.4g/kg/d,静滴,一般连续用 5天为一个疗程。
4. 激素联合其他免疫抑制剂
激素冲击治疗收效不佳时,尤其合并其他自身免疫疾病的患者,可选择激素联合其他免疫抑制剂治疗方案。如联合环磷酰胺治疗,终止病情进展。
缓解期预防性治疗
经过急性期的治疗,NMO 多数都可转入缓解期,突然停药或治疗依从性差都极易导致 NMO 复发。对于急性发作后的复发型 NMO 及NMOSDs 同时合并血清 NMO-IgG 阳性者应早期预防治疗。目前的方案有硫唑嘌呤,吗替麦考酚酯,美罗华,米托蒽醌,环磷酰胺,甲氨蝶呤,静脉注射免疫球蛋白及强的松。硫唑嘌呤、吗替麦考酚酯与利妥昔单抗是最常用的长期预防性药物。
干扰素 [8] 、那他珠单抗 [9] 及芬戈莫德 [10] 可能会使NMO病情加重。
1.硫唑嘌呤
硫唑嘌呤完全起效需4-6个月,在完全起效前可合用小剂量激素。对于AQP4抗体阳性患者应长期应用免疫抑制剂,以防止复发。
用法:按体重 2-3mg/(kg.d)单用或联合口服泼尼松〔按体重1mg/(kg.d)〕,通常在硫唑嘌呤起效后将泼尼松渐减量。对于AQP4抗体阳性患者应长期应用免疫抑制剂,以防止复发。
副作用:发热、恶心、呕吐、白细胞降低、血小板减少、胃肠道、肝功能损害、肌痛、感染、轻度增加罹患肿瘤风险等。在用药治疗初期应每周监测血常规,其后可改为每2周一次,稳定后 1-2月复查一次,并应保证每 2-3个月复查肝功能。
2. 吗替麦考酚酯
通常用于硫唑嘌呤不耐受患者的治疗。1~3g/d,口服。常见的副作用有胃肠道症状和增加感染机会。
3.利妥昔单抗
利妥昔单抗是特异性针对 CD20 的单克隆抗体,能够有效减灭 B 淋巴细胞,从而达到治疗目的。优点是起效快(2周内完全起效),每六个月输液2次。
对症及康复治疗
通过支持治疗,可以使患者的功能障碍得到改善并提高其生活质量。目前,尚无专门针对NMO 的对症支持治疗相关研究发表,大多数治疗经验均来自对 MS 的治疗。
1.痛性痉挛 可应用卡马西平、加巴喷汀、巴氯芬等药物。对比较剧烈的三叉神经痛、神经痛,还可应用普瑞巴林。
2.慢性疼痛、感觉异常等 可用阿米替林、选择性去甲肾上腺素再吸收抑制剂(SNRI)、去甲肾上腺素能和特异性5羟色胺能抗抑郁剂( NaSSA)、普瑞巴林等药物。
3.抑郁焦虑 可应用选择性5羟色胺再吸收抑制剂(SSRI)、SNRI、NaSSA类药物以及心理辅导治疗。
4.乏力、疲劳 可用莫达非尼、金刚烷胺。
5.震颤 可应用盐酸苯海索、盐酸阿罗洛尔等药物。
6.膀胱直肠功能障碍 尿失禁可选用丙咪嗪、奥昔布宁、哌唑嗪等;尿潴留应间歇导尿,便秘可用缓泻药,重者可灌肠。
7.性功能障碍 可应用改善性功能药物等。
8.认知障碍 可应用胆碱酯酶抑制剂等。
9.行走困难 可用中枢性钾通道拮抗剂。
10.下肢痉挛性肌张力增高 可用巴氯芬口服,重者可椎管内给药,也可用肉毒毒素A。
11.肢体功能训练 在应用大剂量糖皮质醇激素时,不应过多活动,以免加重骨质疏松及股骨头负重。当减量到小剂量口服时,可鼓励活动,进行相应的康复训练。
疾病预后
语音
Ghezzi A等人报道NMO年复发率1.3 ± 1.2,发病5年、10年、15年达到EDSS3.0分的比例分别是65%, 82% 及 86%;达到EDSS6.0的比例分别是42%, 53 % 及69% ;达到EDSS10.0的比例分别是8%, 12% 及23%。达到EDSS3.0与发病年龄、第一次发作与第二次发作时间间隔及复发率相关。达到EDSS6.0与发病时基础EDSS分数及复发率相关。复发型 NMO 的相关危险因素包括女性、发病年龄较晚、首次发作脊髓炎时运动障碍不重、伴发自身免疫疾病、疾病最初两年复发频率高。
疾病护理
语音
视神经脊髓炎病程长,易复发,患者及家属应当明白坚持服药的重要性,提高用药的依从性。尽量避免诱发因素,如感冒、发热、感染、生育、外伤、寒冷、拔牙、过劳和精神紧张,不能随意进行疫苗接种,加强肢体功能锻炼以保持活动能力。以心理康复为指导,功能康复为核心,增强患者战胜疾病的信心,从而最大限度地提高患者的生存质量。
心理护理
倾听患者诉说,体会患者的处境和感受,了解其心理状态,使患者树立战胜疾病的信心和勇气,使患者在积极气氛中产生乐观的态度,以积极的情绪接受治疗。
安全护理
NMO患者可能存在功能缺损,如视力障碍,肢体无力等。易发生碰伤、跌伤和坠床等意外。因此,病房内布局要安全合理,光线充足,地面平坦、清洁无积水,无阻碍物,浴室内设有扶手,患者床两侧安放防护架,降低床的高度,不穿拖鞋,穿平底鞋或防滑鞋。
药物护理
遵医嘱按时规律服药,不能随意减量或停用。
饮食护理
避免粗纤维和热烫坚硬食物及刺激性食物,进食低脂、高蛋白、富含维生素及含钾高、钙高的饮食,同时以含丰富亚油酸的食物为宜。多饮水,多食肉类、蔬菜与水果,以增加蛋白质和维生素的摄入。
预防感染护理
①预防肺部感染 瘫痪患者长期卧床,活动量减少,抵抗力低下,极易并发肺部感染,要经常鼓励和协助患者翻身及拍背,2 h/次,拍背时由下向上,2-3 min/次,右侧卧位时拍左侧,左侧卧位时拍右侧,能避免分泌物淤积在下呼吸道,有利分泌物的排出,防止坠积性肺炎的发生。保持口腔清洁,每日至少刷牙或口腔护理2次。保持室内清洁和空气流通,定期空气消毒,房间地面及物品表面用O.5%过氧醋酸擦拭。
②预防褥疮护理 定时翻身,同时按摩肩胛部、骶尾部、足跟、脚踝等骨突处,易受压部位用气圈、棉圈、海绵垫予以保护,经常用温水擦洗背部和臂部,涂爽身粉,保持皮肤清洁。用热水袋时水温不超过50℃,定时按摩,促进血液循环,及时清洗或更换污湿的床褥及衣服,保持床铺平整、清洁,使患者舒适,预防压疮。
③预防尿路感染 对轻度尿潴留者,以温毛巾热敷下腹部并轻度按摩,改变体位,采用习惯的蹲位或直立位小便,听流水声诱导排尿,对诱导排尿失败的患者行导尿。
④预防便秘 由于脊髓损伤,瘫痪卧床,食欲下降,肠蠕动减弱,加之自主神经紊乱易致便秘,应多食水果蔬菜及粗纤维食物,养成定时排便的习惯,可在进食后l-2 h按摩腹部以促进肠蠕动,必要时给予开塞露、温开水或肥皂水灌肠以助排便。
康复护理
应早期帮助患者采取肢体功能锻炼,主要包括体位摆放,定时翻身练习等。利用躯干肌的活动,通过联合反应,共同运动,姿势反射等手段,促使肩胛带的功能恢复,达到独立完成仰卧位到床边坐位的转换,先从大关节开始后到小关节,手法由轻到重,循序渐进恢复肌力,肌力尚可时,鼓励患者积极训练站立和行走,开始扶物训练和久站,逐渐训练独立行走,并可辅以按摩、理疗、针灸,加速神经功能恢复,改善患者的功能状态。
以上内容全部来自《百度百科》
上一篇:风湿类风湿性关节炎
下一篇:没有了
